
Heidelberg Pharma AG


 Thread abonnieren
Thread abonnieren
|
--button_text--
interessant
|
|
witzig
|
|
gut analysiert
|
|
informativ
|
0


Optionen
0
Patients undergoing bone marrow transplant or stem cell gene therapy are first prepared, or conditioned, with non-specific, genotoxic chemotherapy alone or in combination with total body irradiation. This conditioning process is associated with significant toxicity, including development of cancers, infertility, organ toxicities, or even death. As a result, many patients do not consider undergoing a bone marrow transplant or gene therapy. Magenta is developing a portfolio of targeted antibody drug conjugates (ADCs), including non-genotoxic agents, that selectively remove the specific cells to enable a successful transplant or gene therapy procedure.
“This year’s five ASH data presentations from our portfolio of targeted conditioning programs give further insight into our progress in addressing one of the major challenges of the transplant and gene therapy process: the genotoxic conditioning that leaves patients infertile and puts them at risk for malignancies and organ toxicity,” said Michael Cooke, Ph.D., chief scientific officer, Magenta Therapeutics. “Our most advanced targeted conditioning program, C200, is focused on ADCs directed at CD117, a target expressed on stem cells and many types of leukemia cells. Preclinical data at ASH this year show potent and selective depletion of human and non-human primate stem cells with our non-genotoxic CD117 ADC conjugated to amanitin. In addition, both this ADC and our CD45-targeted ADC from our C100 program demonstrated the additional benefit of anti-leukemia activity and survival advantage in patient-derived leukemia models. Based on these promising data, we are finalizing the CD117 and CD45 ADCs, and we expect to select a lead for development for the CD117 program to launch IND-enabling studies in 2019.”
Optionen
0
CD117-Amanitin Antibody Drug Conjugates Effectively Deplete Human and Non-Human Primate HSCs: Proof of Concept as a Targeted Strategy for Conditioning Patients for Bone Marrow Transplant, Abstract #3314
Key results, presented by Brad Pearse, Ph.D., Magenta Therapeutics:
An anti-CD117 ADC conjugated with amanitin potently depleted human and non-human primate hematopoietic stem cells and progenitors in vivo.
An anti-CD117 amanitin ADC with engineered fast half-life showed potent stem cell depletion and rapid clearance, providing appropriate pharmacokinetics for patient preparation for bone marrow transplant.
The ADCs were well tolerated at the efficacious doses.
Potent and selective depletion of stem cells with rapid clearance of the ADC could provide a significant improvement over current approaches to patient preparation with an acceptable safety profile prior to BMT for malignancies, autoimmune diseases and gene therapies, broadening patient access to these potentially curative therapies.
Magenta will next finalize the linker-toxin construct and select a lead for development in 2018, then begin IND-enabling studies in 2019.
Optionen
0
Single Doses of Antibody Drug Conjugates (ADCs) Targeted to CD117 or CD45 Have Potent In Vivo Anti-Leukemia Activity and Survival Benefit in Patient-Derived AML Models, Abstract #3316
Key results, presented by Jennifer Proctor, Magenta Therapeutics:
CD117 is expressed on human hematopoietic stem and progenitor cells and on leukemia cells in 80% of patients with acute myeloid leukemia (AML) and in 65% of patients with myelodysplastic syndromes (MDS); CD45 is expressed on all lympho-hematopoietic cells and in nearly all blood cancers, other than multiple myeloma.
Both the anti-CD117 amanitin ADC (C200 program) and the anti-CD45 amanitin ADC (C100 program) showed potent killing of human hematopoietic stem cells and human leukemia cell lines expressing these targets in vitro.
A single dose of either ADC showed potent in vivo anti-leukemia activity in mice bearing established human leukemia cell lines.
Both ADCs also significantly improved the survival of mice engrafted with human leukemia cells from AML patients, including leukemias that were resistant to multiple lines of therapy including previous allogeneic bone marrow transplant.
Magenta Therapeutics’ C200 and C100 programs are designed with the dual intent of selectively eliminating the necessary cells to enable a successful transplant and reducing disease burden in patients with active disease or in patients who are at high risk of disease relapse.
Magenta will next finalize the linker-toxin constructs, select anti-CD117 and anti-CD45 leads for development and continue to progress ADC-based conditioning approaches targeting CD45 and CD117 toward clinical development.
Optionen
0
Key results will be presented by Rahul Palchaudhuri, Ph.D., Magenta Therapeutics, on Monday, December 3, 2018.
A human/primate cross-reactive anti-CD45 amanitin ADC potently depletes human hematopoietic stem cells and immune cells in culture.
The anti-CD45 ADC achieved efficient depletion of immune cells in the periphery and hematopoietic stem cells in the bone marrow of humanized mice.
Simultaneous depletion of immune and hematopoietic stem cells using an anti-CD45 ADC may enable safer conditioning for allogeneic transplant and enable immune-reset in autoimmune disease transplantation.
Magenta plans to optimize the anti-CD45 ADC and select a development candidate in 2019, with IND-enabling studies to begin in 2020.
Optionen
0
Program: Oral and Poster Abstracts
Type: Oral
Session: 652. Myeloma: Pathophysiology and Pre-Clinical Studies, excluding Therapy: Development of Novel Immunotherapeutic Approaches in Multiple Myeloma
Hematology Disease Topics & Pathways:
antibodies, Diseases, Biological, multiple myeloma, apoptosis, bioengineering, Therapies, Animal models, Biological Processes, Technology and Procedures, Plasma Cell Disorders, Xenograft models, Study Population, Lymphoid Malignancies, Clinically relevant, gene editing, Myeloid Malignancies, flow cytometry, molecular testing, signal transduction
Monday, December 3, 2018: 8:00 AM
Ballroom 20D (San Diego Convention Center)
Ram Kumar Singh, PhD1,2*, Richard J. Jones, Ph.D.2*, Samuel Hong, Ph.D.2*, Fazal Shirazi, PhD2*, Hua Wang, Ph.D.2*, Isere Kuiatse, Ph.D.2*, Andreas Pahl3* and Robert Z. Orlowski, MD, PhD4,5
1Department of Lymphoma and Myeloma, The University of Texas MD Anderson Cancer Center, HOUSTON, TX
2Department of Lymphoma and Myeloma, The University of Texas MD Anderson Cancer Center, Houston, TX
3Heidelberg Pharma AG, Munich, Germany
4The University of Texas M.D. Anderson Cancer Center, Houston, TX
5Department of Lymphoma and Myeloma, MD Anderson Cancer Center, Houston, TX
Background:
Deletion (del) of 17p involving the p53 tumor suppressor (TP53) remains an adverse prognostic factor in multiple myeloma (MM) despite the use of novel agents as well as high-dose chemotherapy with autologous stem cell rescue. Genomic TP53 deletion can cause haploinsufficiency of nearby genes, such as RNA polymerase II subunit A (POLR2A), which ia also located on 17p13.1. We therefore hypothesized that del 17p could reduce POLR2A expression and enhance sensitivity to a-Amanitin, a potent and specific inhibitor of POLR2A and RNA polymerase III.
Methods:
Pre-clinical studies were performed using HDP101, a monoclonal antibody-drug conjugate (ADC) targeting BCMA linked to a-Amanitin, along with unconjugated BCMA antibody, a-Amanitin, and a non-targeting control ADC in myeloma cell line models. The latter included H929, MM1.S, and MOLP-8 TP53 wild-type (WT) lines and isogenic cells in which TP53 had been knocked out (KO) using CRISPR/Cas9 genome editing techniques. To further model del 17p and POLR2A haploinsufficiency, POLR2A expression was knocked down using shRNAs.
Results:
Analysis of the Multiple Myeloma Research Foundation CoMMpassSM database revealed that del 17p13 by SeqFISH was associated with a significant reduction in POLR2A expression by RNASeq (26.05431 fragments per kilobase of transcript per million mapped reads (FPKM) with WT vs. 19.2983 FPKM with del 17p13; p<0.0001). Also, patients within the lower quartile of POLR2A expression, which included those with and without del 17p, had an inferior overall survival (p<0.0011) and a trend towards a worse progression-free survival, suggesting that low POLR2A levels by themselves are an adverse feature. The POLR2A inhibiting anti-BCMA/a-Amanitin conjugate HDP101 induced a time- and dose-dependent reduction in myeloma cell viability post 96-hours of drug exposure starting at concentrations as low as in the picomolar range. This reduction with HDP101 was much greater than that seen with controls, which included a-Amanitin alone, the anti-BCMA antibody without a-Amanitin, or an anti-digoxigenin antibody conjugated to a-Amanitin. When myeloma cells were co-cultured with HS-5 human marrow stromal cells, only a-Amanitin and, to a much greater extent, HDP101 induced a loss of cell viability in myeloma cells, while the stromal cells were spared by HDP101. Loss of cell viability due to HDP101 was associated with induction of apoptosis as judged by the appearance of an increased population of cells that had a sub-G0/G1 DNA content, and that stained positively with Annexin V. Moreover, HDP101 treatment caused the appearance of cleaved fragments of Caspase 9 and 3, and loss of the mitochondrial trans-membrane potential. H929, MM1.S, and MOLP-8 TP53 KO cells were more sensitive to both HDP101 and, less potently to a-Amanitin than were their isogenic TP53 WT parental cells. As H929 cells expressed high levels of BCMA regardless of TP53 or POLR2A status, these were further examined for their sensitivity to HDP101. Notably, the preferential impact upon TP53 KO cells was associated with increased expression of Activating transcription factors -4 and -6, suggesting enhanced induction of endoplasmic reticulum stress, and at least two arms of the unfolded protein response. Interestingly, shRNA-mediated knockdown (KD) of POLR2A expression alone was sufficient to increase baseline levels of apoptosis in both H929 TP53 WT and KO cells, with a greater impact in the latter, supporting the promise of this target. Importantly, KD of POLR2A expression to further model del 17p was also associated with enhanced sensitivity to HDP101 in both the TP53 WT and KO cells compared to the control treatments. Evaluation of HDP101 in primary samples and with in vivo models is underway, and will be presented at the Annual Meeting. These studies were supported by a Leukemia & Lymphoma Society Specialized Center of Research (SCOR-12206-17).
Conclusions:
Our preliminary data support the possibility that del 17p myeloma may have a therapeutic vulnerability to the POLR2A inhibitor a-Amanitin through loss of TP53, and that this sensitivity is further enhanced by decreased POLR2A expression, which is common among del 17p patients. Moreover, they suggest that HDP101 is a novel potent and specific therapeutic that could show enhanced activity in the clinic especially against high-risk multiple myeloma, where effective therapies are still needed to improve patient outcomes.
Disclosures: Pahl: Heidelberg Pharma AG: Employment. Orlowski: BioTheryX, Inc: Consultancy, Membership on an entity's Board of Directors or advisory committees; Genentech: Consultancy; Bristol Myers Squibb: Consultancy; Janssen Pharmaceuticals: Consultancy, Membership on an entity's Board of Directors or advisory committees; Celgene: Consultancy, Membership on an entity's Board of Directors or advisory committees; Millenium Pharmaceuticals: Consultancy, Research Funding; Amgen: Consultancy, Membership on an entity's Board of Directors or advisory committees, Research Funding; Poseida: Research Funding.
Optionen
0
Wenn ich in etwa ein Kursziel in drei Jahren benennen möchte, muss ich auch eine Projektion in etwa drei Jahren machen. Vielleicht dann noch die wahrscheinlichsten Szenarien beschreiben. Der Chart folgt dann auch in der Regel der fundamentalen Entwicklung.
Machen wir doch mal solch eine Projektion auf etwa drei Jahre und dann die weitere Aussicht. Wir gehen mal davon aus, dass HP nicht in irgendeiner Form übernommen und veräußert wird. Fangen wir mal mit den Projektionen an.
Technologiepartner:
HP hat im Augenblick zwei Technologiepartner. Takeda und Magenta. Eine weitere große wurde in einem Artikel angekündigt. Wahrscheinlich werden wir dann im Laufe der nächsten Jahre noch mit ein oder zwei weiteren rechnen können. Ich denke mal, dass die Zahlungen an HP jetzt langsam mit dem weiteren Erfolg steigen werden. Nehmen wir hierfür für alle Partner Optionsmodelle an. Wir hätten dann in drei Jahren etwa vier oder fünf Technologiepartner mit etwa 15-17 Wirkstoffkandidaten. Im Schnitt dürfte der Optionspreis pro Wirkstoff wohl signifikant höher wie etwa 100 Millionen $ liegen. Für die Wirkstoffentwicklung der Partner würden dann in steigender Höhe allein die Optionsbeträge schon bei etwa 2 Milliarden $ liegen, wenn alle Optionen gezogen werden. Danach kämen dann bei fertiger Entwicklung etwa 5% Umsatzbeteiligung über die Laufzeit des Wirkstoffes. Magenta geht nächstes Jahr schon mit CD117 in die Klinik, also die Phase1. Kurz darauf folgt schon CD45. In drei Jahren dürften wohl von der gesamten Pipeline 4-5 Wirkstoffe in der Phase1/2 sein. 5-6 in der vorklinischen Phase und 5-6 in der Forschungsphase. Dies zur Situation der Technologiepartner.
Redectane TLX-250 Girentuximab
Dann aus der alten Pipeline von Wilex Redectane evtl Mesupron und Rencarex. Mesupron und Rencarex lassen wir mal weg, da sie im Augenblick bei den Partnern keine Priorität haben bzw. nicht auslizenziert sind. Bleibt also der Hoffnungsträger Redectane, das an Telix auslizensiert ist. Sie sind hier schon in Australien in der diagnostischen Anwendung in der Phase 3. In Europa wird die Studie wohl in wenigen Tagen oder Wochen folgen. In der Phase 2 folgen drei Studien für die therapeutische Anwendung auch in den nächsten Wochen. Schon etwa Q3/Q42019 sind diese Studien beendet und nach der Auswertung in der diagnostischen Anwendung steht hier der Zulassungsprozess an. In der therapeutischen Anwendung werden dann direkt danach die finalen Studien geplant. Telix treibt diese Studien mit enormen Tempo an. Indikationserweiterung und neue Anwendungsmethoden mit Girentuximab werden erprobt. Die Zulassung hier in der diagnostischen Anwendung etwa 2021. In der therapeutischen Anwendung etwa 2 Jahre später. Allein mit Redectane winken HP über die gesamte Laufzeit Einnahmen von etwa 200-250 Millionen €. Und das ohne Indikationserweiterung und neue Anwendungsgebiete. Der Betrag könnte sich bei positiver Entwicklung über die nächsten 10 Jahre noch einmal auf etwa 400 Millionen € verdoppeln. Das zu Redectane.
Eigene Pipeline:
Dann zu HDP 101 Die technologische Entwicklung scheint nun abgeschlossen und es werden im Dezember Tierversuche gestartet um die optimale Dosis für die Phase 1 zu bestimmen Schon etwa ende 19 befinden wir uns hier in der Phase 1/2. Wenn du die letzte Präsentation zu TP53 und das Abstract von MD Anderson gelesen hast, sieht man das hier ein ganz heißes Eisen im Feuer ist, dass schon in etwa 4-5 Jahren zur Zulassung führen kann. Die Daten scheinen sehr überzeugend zu sein so dass man hier Hoffnung auf eine verkürzte Zulassung hat. Wahrscheinlich werden diese Projekte dann auslizensiert. Dann aber auch mit entsprechenden Anfangszahlungen und Umsatzbeteiligungen. Ich mag die Höhe noch gar nicht schätzen. Doch dürfte man auch hier bei über 1 Milliarde $ liegen bei etwa 15-20% Umsatzbeteiligung. Weitere Kandidaten werden entwickelt, stehen aber noch nicht im Fokus.
Kooperationen:
Dann noch die Entwicklungspartnerschaften mit Nordic Nanovector und Mabvax, bei denen anteilig entwickelt wird.
Pipeline in ca. 3 Jahren:
Dann schauen wir uns doch noch mal in der Zusammenfassung in gut drei Jahren an.
4-5 Entwicklungspartnerschaften 4-5 Phase 1/2, 5-6 vorklinisch, 5-6 in der Forschung
HP Pipeline Phase bei MM und TP53 Projekten mit HDP101. Evtl. schon auslizensiert. 3-4 Projekte vorklinisch
Telix: Redectane Diagnostic schon zugelassen. Redectane Therapeutische Anwendung in der Endphase 3. Weitere Anwendungsgebiete in der Vorklinik.
Kooperationen: 2-3 Projekte vorklinisch oder klinische Phase 1
Dies ist jetzt natürlich das günstigste Szenario. Wahrscheinlich werden auch Rückschläge kommen. Nicht alle Optionen werden gezogen werden. Und wenn du von der Bewertung sprichst bei diesen Werten, dann dürfte diese bei HP bei sehr weit über 1 Milliarde $ liegen. Also Kursen bei über 30 Euro. Kann sein, dass es was später kommt, da einige Projekte verzögert sind. Doch das ist in etwa meine Prognose aufgrund der Einschätzung, die ich oben geschrieben habe.
Optionen
1
1
Mit C100 (CD45) folgt man in der Planung mit etwa einem Jahr Verzögerung.
Präsentation und Quelle zu dem Magenta Programm:
https://investor.magentatx.com/static-files/...427a-b54f-c22d245720fb
Abstracts zu CD117 und CD45:
https://investor.magentatx.com/static-files/...4dd1-a9c9-2773a80cd65a
https://investor.magentatx.com/static-files/...4e89-aab0-5357d47b585a
https://investor.magentatx.com/static-files/...47cc-89df-434da478e94c
Das Schaubild gibt die zeitliche Planung für C100 und C200 wieder.
Optionen
Angehängte Grafik:
aa_achim_aa-magenta_004.png (verkleinert auf 34%)
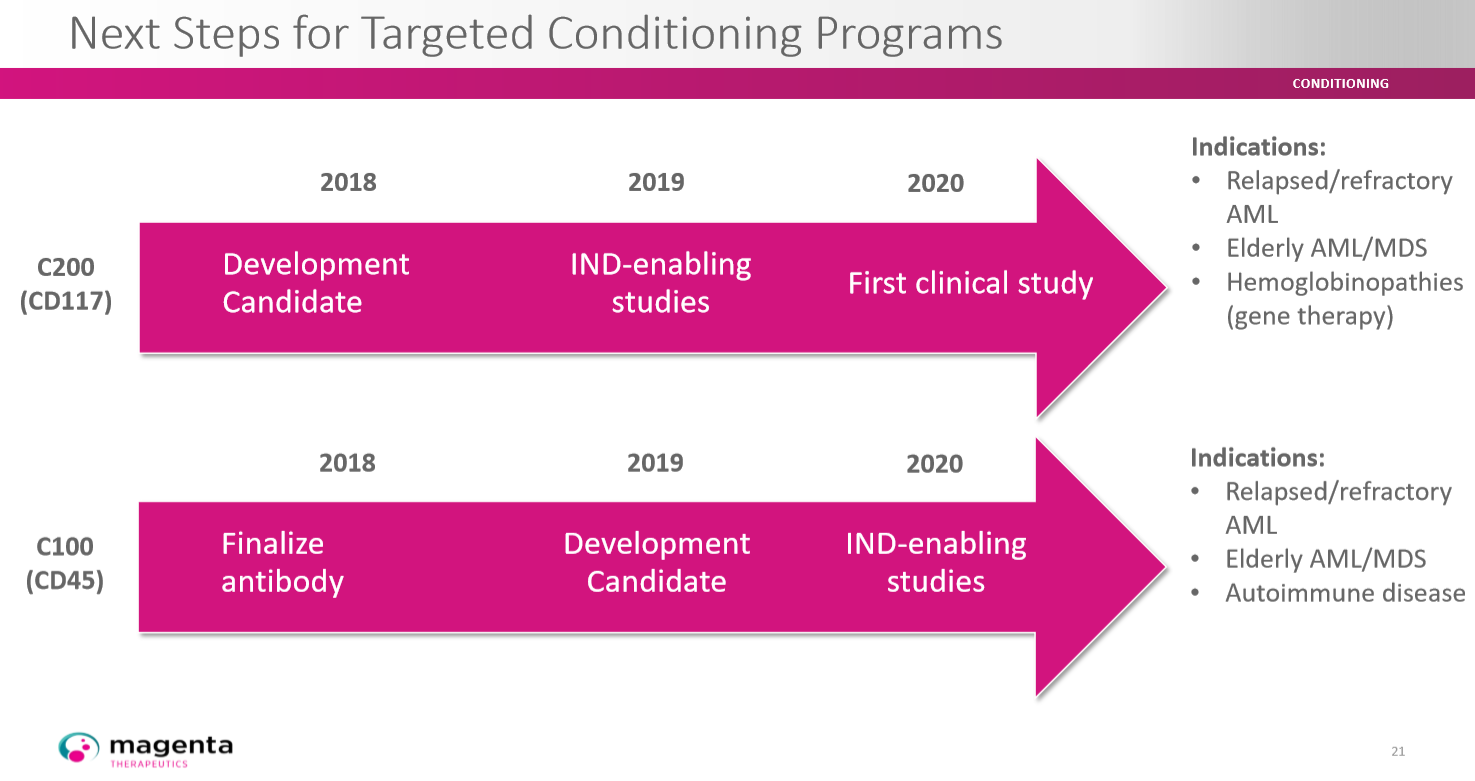
aa_achim_aa-magenta_004.png (verkleinert auf 34%)
0
Redectane-Girentuximab-TLX250
Redectane ist der Wirkstoff, der von HP an Telix auslizensiert wurde. Für jede diagnostische Anwendung erhält HP ca 30% Umsatzbeteiligung. Es kommen jetzt immer mehr Anwendungsgebiete ins Gespräch.
Quantitative imaging of the hypoxia-related marker CAIX in head and neck squamous cell carcinoma xenograft models.
Huizing FJ, Hoeben BA, Franssen GM, Boerman OC, Heskamp S, Bussink J.
Abstract
Introduction Tumor hypoxia plays a major role in radio- and chemotherapy resistance in solid tumors. Carbonic AnhydraseIX (CAIX) is an endogenous hypoxia-related protein, which is associated with poor patient outcome. The quantitative assessment of CAIX expression of tumors may steer cancer treatment by predicting therapy response or patient selection for anti-hypoxia or CAIX-targeted treatment. Recently, the SPECT tracer [111In]In-DTPA-girentuximab-F(ab')2 was developed and validated for targeting CAIX. The aim of this study was to optimize quantitative microSPECT/CT of CAIX expression in vivo in head and neck tumor models. Methods Athymic mice with subcutaneous SCCNij153 and SCCNij202 head and neck squamous cell carcinoma xenografts were injected with [111In]In-DTPA-girentuximab-F(ab')2. First the protein dose, timing and image acquisition settings were optimized. Tracer uptake was determined by quantitative SPECT, ex vivo radioactivity counting, and by autoradiography of tumor sections. The same tumor sections were immunohistochemically stained for CAIX expression and hypoxia. Results Highest tumor-normal-tissue contrast was obtained at 24 hours after injection of the tracer. A protein dose of 10 µg resulted in the highest tumor-to-muscle ratio at 24 hours p.i. Ex vivo biodistribution studies showed a tumor uptake of 3.0 ± 0.6 %ID/g, and a tumor-to-muscle ratio of 8.7 ± 1.4 (SCCNij153). Quantitative analysis of the SPECT images enabled us to distinguish CAIX antigen blocked from non-blocked tumors, fractions positive for CAIX expression: 0.22 ± 0.02 versus 0.08 ± 0.01 (p < 0.01). Immunohistochemical, autoradiographic, and microSPECT/CT analyses showed a distinct intratumoral spatial correlation between localization of the radiotracer and CAIX expression. Conclusion Here we demonstrate that [111In]In-DTPA-girentuximab-F(ab')2 specifically targets CAIX-expressing cells in head and neck cancer xenografts. SPECT imaging with indium labeled girentuximab-F(ab')2 allows quantitative assessment of the fraction of CAIX positive tissue in head and neck cancer xenografts. These results indicate that [111In]In-DTPA-girentuximab-F(ab')2 is a promising tracer to image hypoxia-related CAIX expression
Optionen
0
Quelle: http://www.telixpharma.com/investors/asx-announcements/
Title: ZIR-DOSE Clinical Study Completes Enrolment
Synopsis: The ZIR-DOSE trial is a small 10 patient study that was run as part of the Company’s development of TLX250-CDx. A previous version of the product used 124I (iodine) as the imaging isotope. Iodine is a challenging isotope to work with, is generally expensive to produce and requires additional clinical preparation of the patient (thyroid blocking). Telix elected to replace 124I with new chemistry supporting the use of 89Zr (zirconium). Zirconium easier to use, produces superior quality images and imparts less dose to the patient. The ZIR-DOSE study was run to accurately quantify these improvements and involves administering the product to a small number of patients that are imaged at multiple time-points over the course of a week.
Key points for investors:
• The ZIR-DOSE study further de-risks ongoing product development by accurately measuring the radiation dose to patients from this imaging procedure
• The ZIR-DOSE trial has validated that the dosimetry of TLX250-CDx is within prescribed radiation limits in the product’s intended commercial jurisdictions
• The ZIR-DOSE trial bridges previous iodine-based imaging experience in kidney cancer to Telix’s improved zirconium-based product, confirming significantly improved image quality and radiation characteristics
• The ZIR-DOSE trial confirms the optimal mass dose (injected quantity of imaging agent) for Telix’s ZIRCON multi-centre Phase III trial of TLX250-CDx for the imaging of kidney cancer
ZIR-DOSE Clinical Study Completes Enrolment
Melbourne (Australia), Brussels (Belgium) – 19th December 2018. Telix Pharmaceuticals Limited (ASX.TLX) (“Telix”, the “Company”), a clinical-stage biopharmaceutical company focused on the development of diagnostic and therapeutic products based on targeted radiopharmaceuticals or “molecularly-targeted radiation” (MTR) has today announced that the last patient has been imaged in the ZIR-DOSE bridging study conducted at Radboud University Medical Centre (RUMC) in the Netherlands.
The ZIR-DOSE study is a 10 patient multi-time point imaging study designed to compare the whole-body dosimetry of 89Zr-girentuximab (TLX250-CDx for the imaging of kidney cancer) with historical dosimetry data for 124I-girentuximab (Wilex AG / Heidelberg Pharma). Telix elected to change the isotope from 124I to 89Zr in order to reduce patient dose, enhance image contrast, lower cost of goods and improve clinical workflow by eliminating the need for thyroid blocking. The ZIR-DOSE study achieves both a mass dose comparison between 5mg and 10mg injected dose, as well as full body dosimetry.
Telix has previously reported (ASX release : 15/10/18) that interim analysis of the ZIR-DOSE study indicated a 25% reduction in dose through the change of isotope from 124I to 89Zr, as well as significantly lower liver dose (at 10mg) and superior image quality. The interim analysis has been used to inform the design of the ZIRCON Phase III study and has been incorporated into recent regulatory submissions.
The final analysis of the ZIR-DOSE study will be complete by approximately end-Q1 ’19 but is not expected to differ in outcome from the interim results. Telix co-founder and CEO, Dr. Christian Behrenbruch stated, “The ZIR-DOSE study is a very demanding study for patients because of the number of visits required and the extensive amount of imaging time required to measure dosimetry. The commitment of study participants to contribute to a better way of staging and managing kidney cancer is truly commendable and we are grateful to our partners at RUMC for their efforts in recruiting this trial in a timely fashion.”
Optionen
0
– Japanese Regulatory
Progress Update Melbourne (Australia), Kyoto (Japan)
– 19th December 2018.
Telix Pharmaceuticals Limited (ASX.TLX) (“Telix”, the “Company”), a clinical-stage biopharmaceutical company focused on the development of diagnostic and therapeutic products based on targeted radiopharmaceuticals or “molecularly-targeted radiation” (MTR) has today announced that it has completed first and second stage consultations with the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) and has been granted permission to submit a final stage application by the 11th January 2019 for a Phase I/II study of 89Zr-girentuximab (TLX250-CDx) in Japan.
The objective of the study is to validate the existing US and EU experience of imaging kidney cancer with girentuximab (anti-CAIX antibody) in Japanese patients. The study, as proposed, will build on the EU experience with TLX250-CDx, particularly the ZIR-DOSE study (EudraCT 2017-004769-2). The trial, which is expected to commence in Q2 2019 (subject to regulator approval), will use product manufactured in Japan by Telix’s partner JFE Engineering and will bridge to the global ZIRCON Phase III study (EudraCT 2018-002773-21).
Telix Pharmaceuticals Japan President, Dr. Shintaro Nishimura stated, “Over the past 6 months, Telix has made considerable progress with TLX250-CDx in Japan and our preliminary engagement with the PMDA has been very helpful in terms of developing a roadmap for this product in Japan. The imaging of CAIX in Japanese patients has potential utility in a wide variety of cancers, including renal cancer, and we are pleased with the progress in both manufacturing and regulator engagement.”
Optionen
0
Vielleicht hier noch interessant. Hier wird der Einsatz von Girentuximab-TLX250-Redectane bei einer Vielzahl von Krebsarten angesprochen. Es ist sehr gut möglich das wir hier bald mit Indikationserweiterungen rechnen können. Einige Abstracte und Meldungen von Telix habe ich hier im Thread ja schon gepostet. Demnach wären dann auch für diese Indikationserweiterungen im diagnostischen Bereich Umsatzbeteiligungen in Höhe von etwa 30% an HP fällig. Es dürfte zwar noch etwas dauern. Doch es dürfte die Einnahmen für HP mit Redectane wohl langfristig mehr als verdoppeln. Das Gute an der ganzen Sache. Diese Entwicklung wird von Telix und deren Partnern übernommen und kostet HP keinen Cent mehr. Am Ende des Tages könnte dies für HP zusätzlich von mehreren 100 Millionen $ bedeuten ohne noch einen einzigen Cent zu investieren.
Optionen
0
Abstract:
Heterozygous deletion of chromosome 17p (17p) is one of the most frequent genomic events in human cancers. Beyond the tumor suppressor TP53, the POLR2A gene encoding the catalytic subunit of RNA polymerase II (RNAP2) is also included in a ~20-megabase deletion region of 17p in 63% of metastatic castration-resistant prostate cancer (CRPC). Using a focused CRISPR-Cas9 screen, we discovered that heterozygous loss of 17p confers a selective dependence of CRPC cells on the ubiquitin E3 ligase Ring-Box 1 (RBX1). RBX1 activates POLR2A by the K63-linked ubiquitination and thus elevates the RNAP2-mediated mRNA synthesis. Combined inhibition of RNAP2 and RBX1 profoundly suppress the growth of CRPC in a synergistic manner, which potentiates the therapeutic effectivity of the RNAP2 inhibitor, α-amanitin-based antibody drug conjugate (ADC). Given the limited therapeutic options for CRPC, our findings identify RBX1 as a potentially therapeutic target for treating human CRPC harboring heterozygous deletion of 17p.
Optionen
0
Es gab gestern ein Telix Update. Hier die wichtigsten Eckdaten zu Redectane
Redectane Therapeutisch TLX-250 Phase 2 Q419 Rekrutierung beendet. Daten werden ausgelesen. Umsatzschätzung ca 400-500 Millionen $. Beteiligung HP 5%.
Redectane TLX-250 Diagnostik Phase 3 Q419 Rekrutierung beendet, Daten werden ausgelesen.
Umsatzschätzung 250 Millionen $ Beteiligung HP 30%. Hierzu wird noch vermerkt, das im Augenblick zu diesem Verfahren keine Alternative besteht. Bisher gingen wir von einem Umsatz von ca 70 Millionen $ aus. In der jetzigen Präsentation wird der Jahresumsatz auf ca. 250 Millionen $ geschätzt. Damit müssen wir auch die Beteiligung für HP anpassen. Gingen wir bei der Diagnostik immer von einer Umsatzbeteilgung von ca 20 Millionen $ für HP aus wird sie nach dieser Präsentation bei etwa 75 Millionen $ liegen. Die Studie wird wahrscheinlich dieses Jahr beendet und das Zulassungsverfahren dürfte schon nächstes Jahr gelingen können. Somit kann HP ab ca 2021 mit Einnahmen mit sukzessiven Steigerungen bis zu ca 75 Millionen $ nur mit Redectane TLX-250 aus der Diagnostik geben. Das ist eine riesige Nachricht, zumal hierzu keine andere Alternativen bestehen.
Optionen
1
Redectane-TLX250 oder auch Girentuximab benannt ist von Heidelberg Pharma an Telix auslizensiert.
Es kann in zwei Bereichen der Diagnostik oder auch therapeutisch eingesetzt werden.
In der therapeutischen Anwendung befindet man sich in der Phase 2 und das Ende der Rekrutierung wird etwa Q4 19 erwartet. Sollte die Entwicklung normal verlaufen folgt eine Phase 3 Studie und der Zulassungsprozess. Telix rechnet hier mit Einnahmen von ca 400-500 Millionen$. Erste Einnahmen dürften wir in der herapeutischen Anwendung etwa ab 2023 erwarten. Die Beteiligung für HP beträgt ca 5%.
In der Diagnostik befindet man sich in Phase 3 und das Ende der Rekrutierung wird etwa Q4 19 erwartet. Danach folgt die Auswertung und der Zulassungsprozess. Bitte entnehmt die Einzelheiten der aktuellen Januar Präsentation.
http://www.telixpharma.com/investors/asx-announcements/
Es ist gestern gar nicht so richtig zur Kenntnis genommen worden. Doch die Umsatzschätzung für die diagnostische Anwendung wird auf etwa 250 Millionen $ geschätzt. Dazu kommt dann zusätzlich der gleiche Betrag langfristig wird zusätzliche Indikationen. Die Beteiligung für HP beträgt hier ca 30%. Und man befindet sich nun schon in der Phase 3 . Erste Einnahmen bis zu ca 75 Millionen $ dürften etwa ab 2021 erwartbar sein. Langfristig kommt dann noch der gleiche Betrag für zusätzliche Indikationen dazu, da HP für alle diagnostischen Anwendungen mit Redectane mit etwa 30% beteiligt ist. Das Schaubild bitte ich euch aus anderen bekannten Threads anzuschauen, da hier aus technischen Gründen die Präsentation nicht möglich ist. Es ist aber auch in der Telix Präsentation enthalten. . Dies beschreibt jetzt nur die Chancen die HP mit Redectane hat. Mittelfristig können wir hier mit Einnahmen bis zu 100 Millionen € rechnen . Langfristig dürfte eine Verdoppelung dieser Umsatzbeteiligungen möglich sein. Dies noch mal zu Telix und Redectane-TLX250-Girentuximab.
Optionen
1
Ladenburg, 22. Januar 2019 - Die Heidelberg Pharma AG (FWB: WL6) gab heute bekannt, dass sie von ihrem Kooperationspartner Link Health Co., Guangzhou, China, eine Meilensteinzahlung erhalten hat. Hintergrund ist die Genehmigung der chinesischen Behörde für Lebensmittel- und Arzneimittelsicherheit (National Medical Product Administration - NMPA) für die Durchführung von klinischen Studien mit dem Produktkandidaten MESUPRON(R).
Die Genehmigung erfolgte nach den neuen Regularien der NMPA, welche eine Nutzung der Sicherheitsdaten aus den früheren Studien mit MESUPRON(R) zulassen und damit unmittelbar die Durchführung einer Phase II-Studie möglich machen können. Aufgrund der neuen regulatorischen Situation muss dazu aber eine Überarbeitung des klinischen Studienprotokolls durch Link Health erfolgen. Diese ist in den kommenden Monaten geplant.
Informationen über MESUPRON(R) und das uPA-Programm
Der uPA-Inhibitor MESUPRON(R) (INN: Upamostat) hemmt das Urokinase Plasminogen Aktivator (uPA)-System. 2014 wurden die Rechte zur Entwicklung und Kommerzialisierung von MESUPRON(R) an Link Health Co., Guangzhou, China, für die Region China, Hong Kong, Taiwan und Macao auslizenziert. Grundlage dafür waren Daten aus zwei Phase IIa-Studien in den Indikationen lokal-fortgeschrittener Bauchspeicheldrüsenkrebs (2010) und metastasierter Brustkrebs (2012), die Sicherheit und Aktivität des Medikamentenkandidaten in Kombination mit Chemotherapeutika gezeigt haben. Das Wirkprinzip des MESUPRON(R) könnte einen neuen Behandlungsansatz durch spezifische Blockierung der Metastasierung bei soliden Tumoren bilden.
Optionen
0
HP arbeitet zur Zeit an einem prognostischen Biomarker um die Patientgruppe mit p 53 Merkmalen zu erfassen. Hierzu werden in Abstracts immer mehr Anwendungsgebiete angesprochen. Es scheint ein Arbeitsfeld mit einem sehr beträchlichen Umfang und damit auch Potential für HP und seiner Amanitin Behandlungsmöglichkeit zu sein.
Abstract: https://www.ncbi.nlm.nih.gov/pubmed/30703342
Near-comprehensive resequencing of cancer-associated genes in surgically resected metastatic liver tumors of gastric cancer.
Ikari N1, Serizawa A2, Mitani S3, Yamamoto M2, Furukawa T4.
Author information
Abstract
Liver metastasis is a major cause of death in patients with gastric cancer. We evaluated molecular alterations in clinically resected liver metastases of gastric cancer to identify candidate biomarkers and therapeutic targets. Seventy-four patients, including 37 with liver metastasis that underwent gastrectomy and hepatectomy for gastric cancer and 37 without liver metastasis that underwent gastrectomy for gastric cancer, were studied. Next-generation resequencing was performed for 412 cancer-associated genes in metastatic and/or primary tumors from 30 patients and somatic mutations in TP53, LRP1B, PIK3CA, ADAMTS20, PAX7, FN1, FOXO3, WRN, PTEN, ETV4, and RNF213 were found in metastatic tumors. TP53 mutations were studied by Sanger sequencing in remaining patients; the number of patients with TP53 mutations in metastatic tumors was significantly higher among those with liver metastasis (86.5%, 32/37) versus those without liver metastasis (40.5%; 15/37) (P < 0.0001). TP53 mutations in metastatic liver tumors and corresponding primary tumors were identical in 96.9% (31/32), including some patients with heterogeneous primary tumor components. Immunohistochemical analyses showed aberrant p53 expression in tumors with TP53 mutations. In silico functional evaluations indicated functional loss of missense-mutated TP53. Thus, p53 pathway may facilitate the development of biomarkers and therapeutic approaches to treat gastric cancer metastases to the liver.
Optionen
1
Eine ganze Seite wird Heidelberg Pharma mit den wichtigsten Aspekten gewidmet. Wenn man die Empfehlung so liest meint man, man könnte Heidelberg Pharma 4 oder mehr Seiten widmen, um nur die wichtigsten Aspekte anzusprechen. So vielseitig ist mittlerweile die Pipeline mit erfolgversprechenden Wirkstoffen in Eigenregie aber auch schon an mehrere Partner auslizensiert geworden.
Deshalb empfehle ich allen Lesern hier im Board nach den Fakten ein wenig zu stöbern. Es wird sich bestimmt lohnen. Es ist eine gelungene Vorstellung, der ich aber bedeutend mehr Platz geboten hätte.
Damit erklärt sich auch der Anstieg von Gestern und die gestiegenen Umsätze. €uro ist eine Monatszeitung und wurde erst heute ausgeliefert. Deshalb rechne ich mit einer längeren Phase, an der HP im Fokus steht.